Louvain Drug Research Institute > Cellular and Molecular Pharmacology
|
|
Université
catholique de Louvain Louvain Drug Research Institute > Cellular and Molecular Pharmacology |
|
|
Molecular analysis and pharmacological significance of drug-membrane interactions and membrane destabilization |
|
Quick
links
|
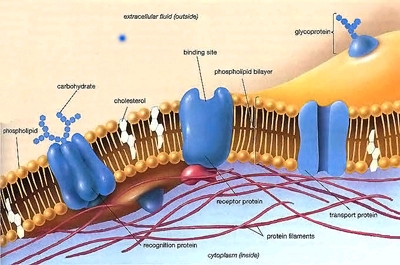
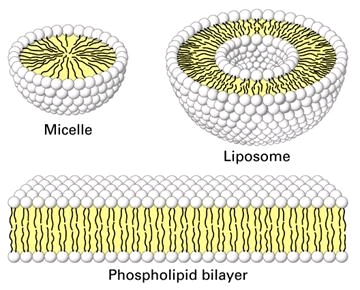
Biological membranes contain lipid bilayers as their basic structural unit. Characterization at the molecular level of the interactions between drugs and lipids help us to understand the critical role of lipids membrane in pharmacology and toxicology. We focus our attention on antibiotics and peptides destabilizing bacterial and cellular membranes. Studies are primarily performed on liposomes (prepared from both pure lipids and lipids extracted from bacterial and other natural sources).These research programs are closely linked to those exploring the cellular toxicity of antibiotics and the novel antibiotic targets . |
| |
|
Team
- Senior investigators: M.P. Mingeot-Leclercq, F. Van Bambeke, P.M. Tulkens
- Doctoral fellows: H. Bensikaddour (2006- ), J. Lorent (2007- )
- Former investigators: J.P. Montenez (1991-1995), C. Kotretsou (1992), A. Kerkhofs (1997-1998), M. Bensliman (1999-2001), D. Tyteca (1997-2002), N. Fa (2003-2006), O. Domenech (2007-2009), M. Ouberaï (2008-2009)
Collaborations
- P.J. Courtoy & D. Tyteca (Endocytosis and epithelial differentiation, de Duve Institute & Université catholique de Louvain, Brussels).
- A. Schanck and K. Snoussi (Institut de la matière condensée et des nanosciences, Secteur des sciences et technologies, Université catholique de Louvain, Louvain-la-Neuve)
- Y.Dufrêne (Bio- and Soft Matter, Institute of Condensed Matter and Nanoscience, Université catholique de Louvain, Louvain-la-Neuve)
- R. Brasseur, L. Lins & M. Deleu (Centre de biophysique moléculaire numérique, Faculté des Sciences Agronomiques de Gembloux, Gembloux Agro Biotech, Gembloux)
- M. Fillet (Laboratory of Medical Chemistry, GIGA, Université de Liège, Liège)
- E. Goormaghtigh (Structure et Fonction des Membranes biologiques, Université Libre de Bruxelles, Brussels)
- J.L. Decout (Département de Pharmacochimie Moléculaire, Université de Grenoble, Grenoble, France)
Main current research programs
- Macrolides: potential mechanism of inhibition of fluid phase endocytosis
- Fluoroquinolones: potential mechanims of differential accumulation by macrophages
- Lysostaphin, lipopeptides and lipoglycopeptides: bacterial envelope as a target for antibacterial effect
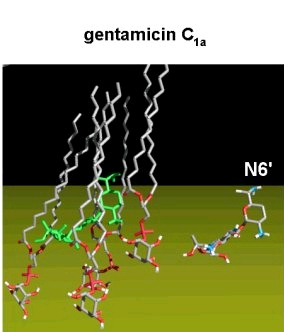
- Amphiphilic derivatives of aminoglycosides : study of their mode of action
Cell membranes often constitute the first biological structure encountered by drugs. Our laboratory has specialized in the biophysical and biochemical studies of the interactions between drugs and membrane lipids.
|
The main questions we address are
Interactions between azithromycin and lipids : potential role for inhibition of fluid phase endocytosis
In J774 macrophages and fibroblasts, the macrolide antibiotic azithromycin inhibits fluid-phase endocytosis (studied with horseradish peroxidase and lucifer yellow), and delays the sequestration of receptor-bound transferrin and peroxidase-anti-peroxidase immune complexes into cell-surface endocytic pits and vesicles. This suggests that azithromycin alters membrane dynamics, which we have studied with membrane models.
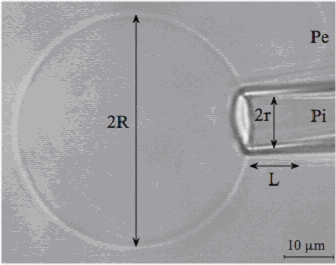
Using the micropipette aspiration technique on giant unilamellar vesicle (GUVs) made of dioleoylphosphatidylcholine (DOPC) (see figure 1A), we showed that azithromycin alters elastic moduli (decrease in bending modulus and apparent area compressibility modulus). Insertion of azithromycin into the DOPC bilayer reduces the requirement level of energy for thermal fluctuations,as well as the stress to stretch the bilayer. All together, these effects explain the decrease of cohesion and a facilitated merging of dipalmitoylphosphatidylcholine (DPPC) into a DOPC fluid matrix, as observed in atomic force microscopy (AFM). In contrast, azithromycin does not perturb the lipid phases and lipid membrane organization in sphingomyelin (SM) or sphingomyelin:cholesterol (SM:CHOL) domains, which may contribute to explain why azithromycin decreases cellular lipid membrane dynamics (Figure 1B).
Further studies, including determination of elastic moduli on GUVs containing cholesterol and sphingomyelin, have to be perfomed for a better understanding of relevant effects of azithromycin on endocytosis.
|
Selected References on biophysical studies with macrolides (by reverse chronological order; for a full reference list, see our publication list)
Studies with membrane models:
- Fa N., L. Lins, P. Courtoy, Y. Dufrêne, P. Van Der Smissen, R. Brasseur, D. Tyteca & M.-P. Mingeot-Leclercq. 2007. Decrease of elastic moduli of DOPC bilayers by a macrolide antibiotic, azithromycin. Biochem. Biophys. Acta (Biomembranes). 1768: 1830-1838. (PDF)
- Fa N., S. Ronkart, A. Schanck, M. Deleu, A. Gaigneaux, E. Goormaghtigh & M.-P. Mingeot-Leclercq. 2006. Effect of the antibiotic azithromycin on thermotropic behavior of DOPC or DPPC bilayers. Chem. Phys. Lipids. 144:108-116. (PDF)
- Berquand A., N. Fa, Y.F. Dufrêne & M.-P. Mingeot-Leclercq. 2005. Interaction of the macrolide antibiotic, azithromycin, with lipid bilayers: effect on membrane organisation, fluidity and permeability. Pharm Res. 22: 465-475. (PDF)
- Berquand A., M.-P. Mingeot-Leclercq & Y. Dufrêne. 2004. Real-time imaging of drug-membrane interactions by atomic force microscopy. Biochem. Biophys. Acta (Biomembranes). 1664: 198-205. (PDF)
- Tyteca D., A. Schanck, Y. Dufrêne, M. Deleu, P.J. Courtoy, P.M. Tulkens & M.-P. Mingeot-Leclercq. 2003. The macrolide antibiotic azihromycin interacts with lipids and affects membrane organisation and fluidity: studies on Langmuir-Blodgett monolayers, liposomes and J774 macrophages. J. Memb. Biol. 192: 203-215.
Studies with cultured cells:
- Tyteca D., P. Van Der Smissen, M. Mettlen, F. Van Bambeke, P.M. Tulkens, M.-P. Mingeot-Leclercq & P.J. Courtoy. 2002. Azithromycin, a lysosomotropic antibiotic, has distinct effects on fluid-phase and receptor-mediated endocytosis, but does not impair phagocytosis in J774 macrophages. Exp. Cell Res. 281:86-100. (PDF)
- Tyteca D., P. Van Der Smissen, F. Van Bambeke, K. Leys, P.M. Tulkens, P.J. Courtoy, & M.-P. Mingeot-Leclercq. 2001. Azithromycin, a lysosomotropic antibiotic, impairs fluid-phase pinocytosis in cultured fibroblasts. Eur.J.Cell Biol. 80:466-478. (PDF)
Interaction with lipids and potential mechanims of differential accumulation by macrophages
Ciprofloxacin and moxifloxacin, two fluoroquinolone antibiotics, accumulate to different levels and at different rates in J774 mouse macrophages. This may result from their differential recognition by the Mrp4 efflux transporter expressed by these cells (see more details in the section on antibiotic efflux) and/or from a different ability to interact with membrane lipids.
We have therefore investigated the interactions of these two fluoroquinolones with lipids using an array of complementary techniques.
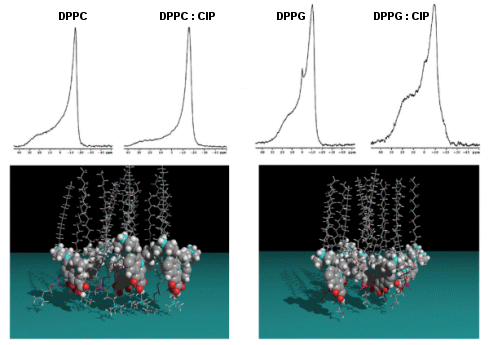
Using Atomic Force Microscopy (AFM; see Figure 2A) and Langmuir studies (measure of surface pressure-area isotherms of monolayers at a air/water interface), and working on membrane models made of mixtures of dioleoylphosphatidylcholine (DOPC) and dipalmitoylphosphatidylglycerol (DPPC), we showed that moxifloxacin induces an erosion of the DPPC domains in the DOPC fluid phase to a greater extent than ciprofloxacin and caused a more important shift of the surface pressure-area isotherms toward lower area per molecule. These effects are related to a lower release of moxifloxacin from lipids to aqueous phase (phase transfer studies and conformational analysis) and a marked decrease in the packing (all-trans conformation) of acyl-lipid chains of DPPC, as shown by Attenuated Total Reflection Fourier transform infra-red (ATR-FTIR).Using quasi-elastic light scattering, steady-state fluorescence anisotropy, ATR-FTIR, 31P NMR, and conformational analysis (Figure 2B), we have also shown that the interaction of ciprofloxacin with lipids markedly depends on the nature of their phosphate head groups, with ciprofloxacin interacting preferentially with anionic lipids like phosphatidylglycerol (PG).
Chronic exposure of J774 macrophages select for cells that overexpress Mrp4. To investigate whether this overexpression is related to changes in lipidic composition of membranes, we have extracted lipids from wild-type cells (basal level of expression of Mrp4) and cells overexpressing Mrp4. We are now comparing them by Mass Spectrometry analyses (qualitative and quantitative studies; collaboration with M. Fillet, Liège) and investigating the interactions of ciprofloxacin with liposomes prepared with this lipids..
|
Selected References on biophysical studies with fluoroquinolones (by reverse chronological order; for a full reference list, see our publication list)
- studies with liposomes:
- Bensikaddour H., N. Fa, I. Burton, M. Deleu, L. Lins, A. Schanck, R. Brasseur, Y.F. Dufrêne, E. Goormaghtigh, & M.-P. Mingeot-Leclercq. 2008a. Characterization of the interactions between fluoroquinolone antibiotics and lipids: a multitechnique approach. Biophys. J. 94: 3035-3046. (PDF)
- Bensikaddour H., K. Snoussi, L. Lins, F. Van Bambeke, P.M. Tulkens, R. Brasseur, E. Goormaghtigh & M.-P.Mingeot-Leclercq. 2008b. Interactions of Ciprofloxacin with DPPC and DPPG: Fluorescence Anisotropy, ATR-FTIR and 31P NMR Spectroscopies and Conformational Analysis. Biochem. Biophys. Acta (Biomembranes). 1778: 2535-2543. (PDF)
- studies with cells (see further details on these studies in the Antibiotic efflux section)
- Marquez B, NE Caceres, M.-P. Mingeot-Leclercq, PM Tulkens, & F. Van Bambeke. 2009. Identification of the efflux transporter of the fluoroquinolone antibiotic ciprofloxacin in murine macrophages: studies with ciprofloxacin-resistant cells. Antimicrob Agents Chemother. 53:2410-2416. (PDF)
- Michot J.M., M.F. Heremans, N.E. Caceres, M.-P. Mingeot-Leclercq, P. M. Tulkens & F. Van Bambeke. 2006. Cellular accumulation and activity of quinolones in ciprofloxacin-resistant J774 macrophages. Antimicrob. Agents Chemother. 50: 1689-1695. (PDF) ·
Antibiotics targeting bacterial envelope (cell wall and membrane lipids)
Potential role in antibacterial effect
The bacterial envelope appears as a promizing target for new antibiotics because (a) its destabilization leads to rapid bacterial cell death and (b) its composition is so critical for life that resistance is unlikely to rapidly develop. We are studying the interaction between drugs and cell wall and/or membrane to decipher their mechanism of action at the molecular levels, which may help in the design of more powerful antibiotics. We are mainly interested in investigating drugs active on Staphylococcus aureus (Gram-postive organism) and Pseudomonas aeruginosa (Gram-negative organism), because of the alarming increase of multiresistance to currently available antibiotics in these bacterial species.
Lysostaphin
Lysostaphin is an enzyme secreted by Staphylococcus simulans, which cleaves the peptidoglycan cross-linking pentaglycin bridges of Staphylococcus aureus. We have studied its interaction with cell wall and have shown that is causes major structural changes (cell swelling, splitting of the septum and creation of nanoscale perforation) that can alter cell wall nanomechanical properties.
Lipopeptides
Surfactin is a lipopeptide produced by Bacillus subtilis, which is highly bactericidal. In situ AFM imaging shows that surfactin is able to solubilize dioleoylphosphatidylcholine / dipalmitoylphosphatidylcholine (DOPC/DPPC) mixture, which could explain is capacity to rapidly kill bacteria. Synthetic analogs of surfactin have been produced which differ in their membrane effects, suggesting a role of lipopetide geometry, charge, and hydrophobicity in these interactions.
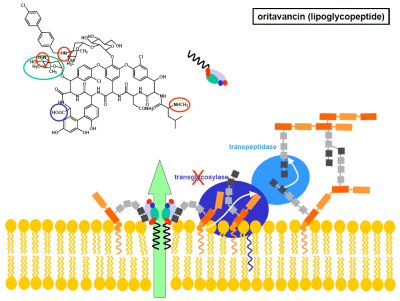
Lipoglycopeptides (Oritavancin)
Glycopeptides bind with high affinity and specificity to peptidoglycan precursors, thereby preventing their incorporation into the bacterial cell wall. hemisynthetic derivatives have been produced, generating a new generation of molecules that all possess a lipophilic side chain and are called collectively lipoglycopeptides. These molecules show improved activity against multi-resistant strains, and, in contrast to vancomyxin, are highly bactericidal, suggesting a novel mode of action that could involve membrane destabilization (Figure 3A).
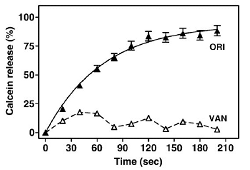
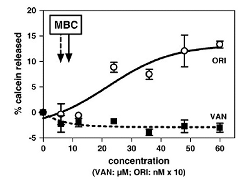
We have compared the capacity of vancomycin and oritavancin to permeabilize lipid bilayers by following the release of calcein entrapped in liposomes at self-quenching concentrations (Figure 3B and C). The results were correlated with real-time observations of the interactions of both molecules with lipids at the nanoscale level using Atomic Force Microscopy (AFM), which showed that oritavancin induces holes and erosion of edges, and decreases the thickness of supported lipid bilayers.
|
Selected References on biophysical studies with lipopeptides and glycolipopeptides (by reverse chronological order; for a full reference list, see our publication list)
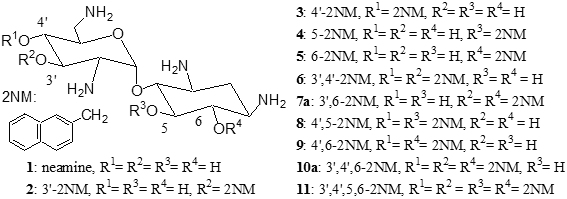
New amphiphilic derivatives of aminoglycosides
Aminoglycosides bind to the 16S bacterial rRNA and disturb the protein synthesis. We have examined the mode of action of amphipathic derivatives of aminoglycosides designed to target bacterial membranes (synthesis made at the Université de Grenoble, France). These are derived from neamine, in which 1 to 4 hydroxyl functions were capped with phenyl, naphthyl, pyridyl, or quinolyl rings. The 3',4'-, 3',6-, and the 3',4',6-2-naphthylmethylene derivatives are active against both sensitive and resistant Staphylococcus aureus strains and the last compound also shows marked antibacterial activity against Gram (-) bacteria, including strains expressing enzymes modifying aminoglycosides, efflux pumps, or rRNA methylases.
We observed that the 3',4',6-2-naphthylmethylene derivative shows only a weak and aspecific binding to a model bacterial 16S rRNA as well as a lower ability to decrease 3H leucine incorporation into proteins in Pseudomonas aeruginosa, suggesting it acts against Gram (-) bacteria through a mechanism different from inhibition of protein synthesis, probably involving membrane destabilization.
Further characterization at the molecular level of the mechanism of action of these molecules are ongoing, to help synthesizing new derivatives active against P. aeruginosa, including resistant strains, and targeting specifically bacterial membranes.
|
Selected References on amphiphilic derivatives of aminoglycosides (by reverse chronological order; for a full reference list, see our publication list) (for a full reference list, consult the publication list)
Additional
information: <tulkens@facm.ucl.ac.be>
Last significant update: January, 2011